SICKLE CELL DISEASE
What is it ?
Photo credits: AFP
Sickle cell disease, also known as sickle cell anemia, is a chronic, genetic, inherited disease caused by a mutation on chromosome 11. It is characterized by an abnormality of hemoglobin, the main protein of the red blood cell. The red blood cells of sickle cell patients have a tendency, especially in cases of hypoxia, to form polymers that deform the red blood cells (S-shaped or sickle-shaped) and clump together, disrupting blood flow, causing extremely painful vaso-occlusive crises and promoting infections. The difference between hemoglobin S and normal hemoglobin A is that normal hemoglobin is soluble when it has oxygen, and it is also soluble when it does not.
Hemoglobin transports oxygen from the lungs to the tissues and is involved in the removal of carbon dioxide. In sickle cell disease, the abnormal shape adopted by the hematite makes them fragile and rigid. The spleen does not recognize them, so it destroys them. These abnormalities therefore promote anemia. In addition, micro clots form and can become larger and larger and the real lethality of sickle cell disease is not due to the anemia, but to this vascular disease. About one-third of people homozygous for hemoglobin S have one or more strokes by the age of 10. And in the lucky few who make it into their teens, these clots can accumulate in the lungs and cause a very serious disease called acute chest syndrome, which is essentially emphysema or the destruction of the lungs by these blockages. About one-third of patients will live healthy into adulthood, but they have many health problems and their lifespan is considered shorter. It is now more than 40 years, whereas it was less than 20 years before the 1980s. However, much remains to be done to improve the daily life and life expectancy of patients.
Mode of transmission
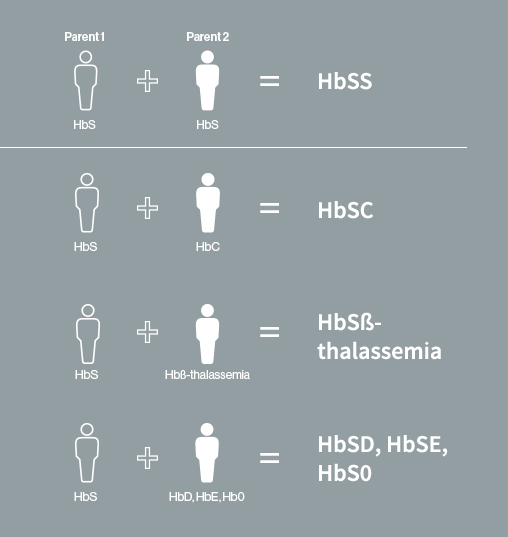
The disease is transmitted in an autosomal recessive manner: each parent must pass on the mutated gene to the child in order for the disease to occur in the child.
See diagram
There are several types of sickle cell disease. The most common form, responsible for the most severe cases, is associated with the presence of two copies of the hemoglobin S gene, one on each chromosome 11 (homozygous form SS). In other forms of the disease, patients carry only one copy of the gene coding for hemoglobin S, the second copy carries a different mutation, either C, which leads to the double heterozygous form SC for example, or others such as thalassemias –
see diagram
Origin
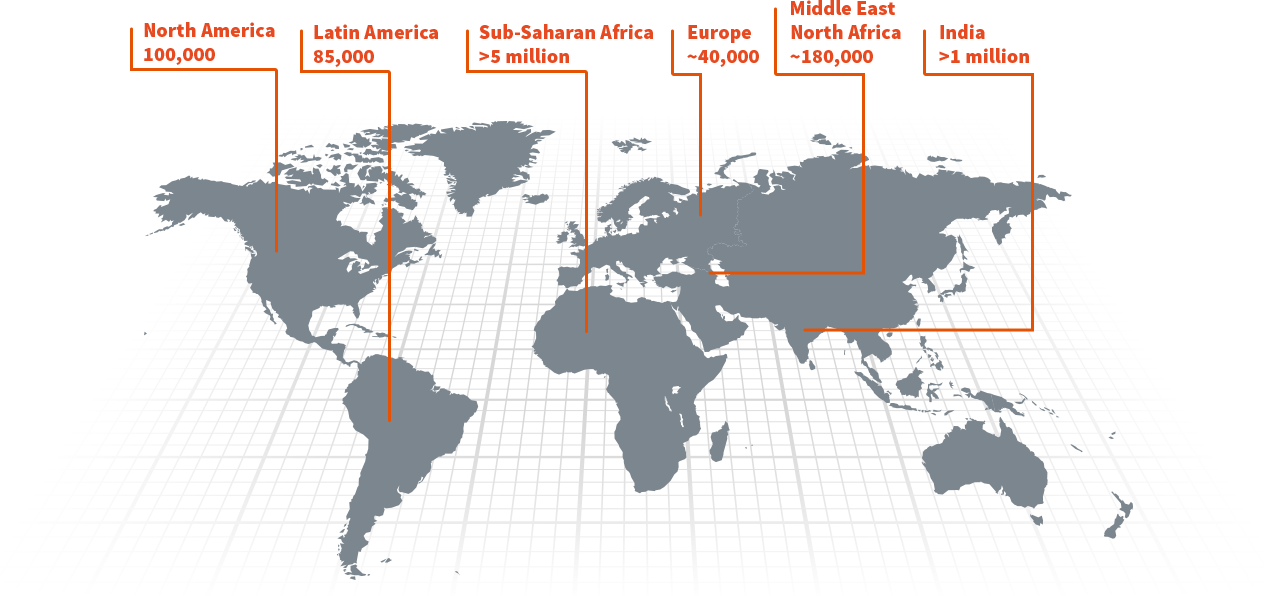
Being of African origin, it mainly affects people of African descent. The mutation would have been “motivated” by a genetic reaction to protect against malaria which was rampant in certain areas. It was thus established that the sickle cell trait protected against malaria. As a result of migration and interbreeding, this gene has moved out of its “comfort zone” and has since spread to become the most common genetic disease in the world. Sickle cell disease is very present in India, America, especially in the United States, the Caribbean and Brazil, as well as in Western Europe. Therefore, people of Mediterranean, Middle Eastern, Caucasian, Indian, Hispanic, Native American descent, among others, may also be affected.
Red blood cell of a patient with sickle cell disease. The origin of sickle cell anemia comes from the modification of a single amino acid, which causes a loss of membrane elasticity and produces hemolysis. Inserm, C. Féo
. The hemoglobin of sickle cell patients is called “hemoglobin S”, for Sickle.
Highly variable and often unpredictable symptoms
Schema
The symptoms of the disease are variable and depend not only on age, but also on the severity of sickle cell disease.
In the very first months of life, infants are generally asymptomatic because they benefit from the presence of fetal hemoglobin, the form of hemoglobin produced in the fetus during the in utero period, which is not mutated. Unfortunately, it then disappears and is replaced by hemoglobin S. Thereafter, patients have an increased risk of developing anemia or sometimes serious infections (pneumonia, meningitis or septicemia…). With advancing age, these risks of complications persist and others appear, more specific, linked to the long-term evolution of the disease.
Anemia
Anemia is often the first sign of the disease. It is manifested by pallor and chronic fatigue, sometimes by jaundice.
The chronic anemia associated with sickle cell disease is caused by hemoglobin S polymers that weaken red blood cells and promote their early destruction (hemolysis). While the average lifespan of red blood cells is normally 120 days, it drops to only about 20 days in people with sickle cell disease.
Anemia can worsen abruptly if the spleen is overactive in destroying abnormal red blood cells. This is called acute splenic sequestration.
Vaso-occlusive crises
Vaso-occlusive attacks are triggered by red blood cells stiffened by hemoglobin S polymers, which obstruct the blood flow in small blood vessels. These attacks cause acute pain, often extremely violent, and affect the bones, joints of the arms and legs, back or chest. In very young children, the attack is usually manifested by painful swelling of the hands and feet (hand-foot syndrome).
Vascular occlusions associated with sickle cell disease can lead to major complications. Thus, acute chest syndrome is a frequent complication in the days following the onset of a vaso-occlusive crisis. It is even the first cause of death in patients with sickle cell disease. In this case, the vaso-occlusion affects the lung and compromises the oxygenation of the whole body. This results in breathing difficulties and chest pain, sometimes accompanied by fever.
Strokes are also common in sickle cell patients, especially in children. Finally, repeated vaso-occlusions can lead to necrosis of certain tissues such as bone (osteonecrosis of the femoral head) or organs. Moreover, the spleen of sickle cell patients, which is very much in demand to ensure the hemolysis of sickle cells, is thus one of the organs that is damaged at an early stage by vaso-occlusions. It no longer fulfils its role completely, which can favour the occurrence of bacterial infections.
Sensitivity to infections
Infections exacerbate other manifestations of sickle cell disease such as anemia and vascular occlusions. In addition, they always represent a risk of mortality (fulminant sepsis), especially in children with weakened defense mechanisms.
Long-term complications
Over the years, all these manifestations of the disease put a strain on the body. They are likely to affect almost all organs, in particular the kidney (renal failure), the osteoarticular system (osteoarthritis, osteoporosis), the eye (intraocular haemorrhages), the liver, the lungs (pulmonary arterial hypertension) or the gallbladder (stones).
Diagnosis as early as possible
This diagnosis is made primarily on the basis of a simple blood sample analyzed by various methods (electrophoresis, isoelectrofocusing or chromatography) capable of distinguishing hemoglobin S from normal hemoglobin A and other hemoglobin variants, based on their physicochemical differences. Advances in the management of the disease have significantly increased the average life expectancy of people with sickle cell disease.
Neonatal screening
France deploys systematic neonatal screening for all children born to parents from the populations most affected by the disease. It is carried out in the maternity hospital at the 72nd hour of the infant’s life (within the broader framework of screening for rare diseases), using a blood sample taken from the heel of the newborn.
Active prevention and symptomatic treatment
For most patients, the management of sickle cell disease is based on the prevention of complications and regular medical follow-up. Neonatal screening is an essential tool.
Prevention of complications
Prophylaxis of the risk of infection consists of preventive administration of antibiotics and reinforcement of the vaccination program, especially in infants and young children. Folic acid and iron supplements are also prescribed to strengthen red blood cell production and prevent anemia.
A healthy lifestyle
These good habits will consist of :
- Adopting a balanced diet is essential.
- Good hydration is necessary (drink plenty of water).
- Avoid exposure to extreme temperatures or significant temperature variations.
- Avoid intense efforts related to the practice of extreme sports and endurance are to be avoided.
- Avoid poorly ventilated rooms and stays at altitudes of over 1,500 meters where oxygen is scarce.
- Wear loose-fitting clothing that does not cut off blood circulation.
- Regular medical follow-up allows to evaluate the evolution of the disease and the damage it causes on the organs.
- Particular attention should be paid to the renal and respiratory functions as well as the state of the eye.
- A cerebral ultrasound (transcranial Doppler) is regularly performed to prevent a risk of stroke.
Blood transfusions
Blood transfusion is an important tool in the management of sickle cell patients. It consists in transfusing the patient with blood from a compatible healthy donor, thus allowing to restore an acceptable level of red blood cells in case of aggravated anemia and to “dilute” sickle cell red blood cells with normal red blood cells.
However, repeated transfusions can lead to red cell alloimmunization: the patient’s immune system reacts against the donor blood, which is considered foreign.
Management of painful attacks
In case of a vaso-occlusive crisis, analgesics relieve the pain. If the pain persists, hospitalization is necessary. The strength of the analgesics will then be increased until morphine or opioid derivatives are administered for the most resistant pain. Painkillers are sometimes supplemented by oxygen therapy.
To reduce the occurrence of painful attacks, hydroxycarbamide (or hydroxyurea) can be prescribed. This drug acts at several levels in the prevention of vaso-occlusions. In particular, it increases the production of fetal hemoglobin, which is free of mutations and present in low concentrations in adults, and reduces the polymerization of hemoglobin S. This treatment has greatly improved the quality of life of sickle cell patients. However, its efficacy may decrease with age and not all adults respond to it. In addition, hydroxycarbamide can lead to (reversible) fertility problems in men.
Bone marrow transplantation
The only curative treatment currently available for sickle cell disease is a bone marrow transplant. This marrow contains the stem cells that give rise to blood cells, including red blood cells. In concrete terms, the patient’s own marrow stem cells are destroyed and replaced by those of a compatible healthy donor, often a brother or sister. This procedure has a 95% success rate and it is possible to find a compatible donor in 70% of cases. However, it remains very cumbersome and costly. It is therefore reserved for the most severe forms of the disease, particularly in children. In France, about twenty patients benefit from such a transplant each year.
Research without borders
Research on sickle cell disease relies heavily on collaboration with the regions most affected by the disease. Researchers are also committed to developing cooperation with low-income countries where most sickle cell patients are concentrated. A better understanding of the disease and its pathophysiology through the epidemiology of cardiovascular complications is, for example, the objective of the CADRE project developed by INSERM researchers (Brigitte Ranque and Xavier Jouven) and GR-Ex laboratory of excellence on red blood cells in several African countries (Cameroon, Senegal, Mali, Ivory Coast and the Democratic Republic of Congo), there are other examples…
Many people are affected by sickle cell disease
Although current treatments have greatly increased the life expectancy of affected patients, they are still limited. Pain management during attacks remains a real problem for patients. Research is therefore continuing its efforts to improve quality of life and even treat the disease at its source, where the red blood cells are born.
Avenues opened up by genetics and epigenetics
The hopes of gene therapy
Because of the difficulty of bone marrow transplants and the difficulty of finding compatible donors, the prospects of curing sickle cell disease are more likely to be based on gene therapy. This approach aims to “graft” a healthy beta-globin gene into the hematopoietic stem cells of sickle cell patients.
Sources:
https://www.inserm.fr/dossier/drepanocytose/
https://robertdebre.aphp.fr/drepanocytose/
https://www.notaloneinsicklecell.com/fr/Global-Impact-Of-SCD/
https://www.genome.gov/genetics-glossary/Sickle-Cell-Disease
https://www.sparksicklecellchange.com
https://www.rarediseaseadvisor.com
www.notaloneinsicklecell.com
NotAloneInSickleCell.com
Click on the image to enlarge
















